Overview
The Asokan Lab engineers biological macromolecules and viruses for human gene therapy.
Our vision has evolved over the years from tackling complexity to seeking simplicity - from studying and engineering Adeno-Associated Viruses (AAV) to back-splicing and trans-splicing RNAs and most recently, designing new immunomodulatory and effector proteins.
We pursue applications across a range of organs, tissues and cell types and our overarching goal is to amalgamate these platform technologies for clinical translation of genetic medicines.
Contact Us
The Asokan lab is actively seeking postdoctoral fellows, graduate students, and research technicians. Contact us at asokanlab@duke.edu if you are interested in joining.
Collaborators
Are you interested in collaborating? We support team science and would love to hear about your work and how we can engage with you. Please email asokanlab@duke.edu.
Publications and Funded Projects
View Dr. Asokan’s profile to see publications and funded projects.
Research
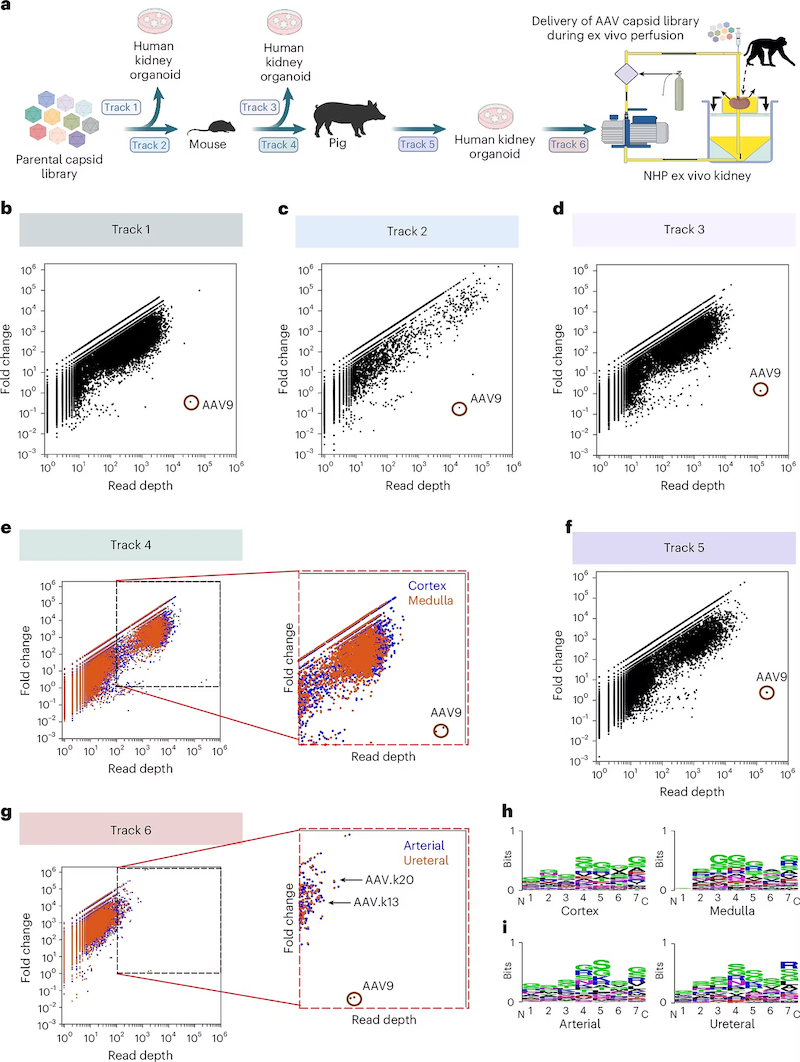
Abstract
The difficulty of delivering genes to the kidney has limited the translation of genetic medicines, particularly for the more than 10% of the global population with chronic kidney disease. Here we show that new variants of adeno-associated viruses (AAVs) displaying robust and widespread transduction in the kidneys of mice, pigs and non-human-primates can be obtained by evolving capsid libraries via cross-species cycling in different kidney models. Specifically, the new variants, AAV.k13 and AAV.k20, were enriched from the libraries following sequential intravenous cycling through mouse and pig kidneys, ex vivo cycling in human organoid cultures, and ex vivo machine perfusion in isolated kidneys from rhesus macaques. The two variants transduced murine kidneys following intravenous administration, with selective tropism for proximal tubules, and led to markedly higher transgene expression than parental AAV9 vectors in proximal tubule epithelial cells within human organoid cultures and in autotransplanted pig kidneys. Following ureteral delivery, AAV.k20 efficiently transduced kidneys in pigs and macaques. The AAV.k13 and AAV.k20 variants are promising vectors for therapeutic gene-transfer applications in kidney diseases and transplantation.
Full Article: https://www.nature.com/articles/s41551-024-01341-0
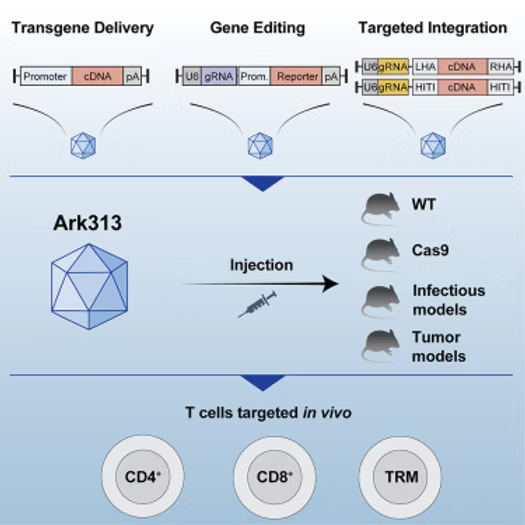
Summary
Genetic engineering of T cells in mouse models is essential for investigating immune mechanisms. We aimed to develop an approach to manipulate T cells in vivo using an evolved adeno-associated virus (AAV) capsid named Ark313. Delivery of a transient transgene expression cassette was feasible using Ark313, and this serotype outperformed natural serotypes. A single intravenous injection of a Cre recombinase-expressing Ark313 in the Ai9 fluorescent reporter mouse model achieved permanent genetic modifications of T cells. Ark313 facilitated in vivo gene editing in both tissue-resident and splenic T cells and validation of immunotherapy targets in solid tumor models. Ark313 delivered large DNA donor templates to T cells in vivo and integrated transgenes in primary CD4+ and CD8+ T cells, including naive T cells. Ark313-mediated transgene delivery presents an efficient approach to target mouse T cells in vivo and a resource for the interrogation of T cell biology and for immunotherapy applications.
Full Text: https://www.sciencedirect.com/science/article/pii/S1074761325000342?via%3Dihub
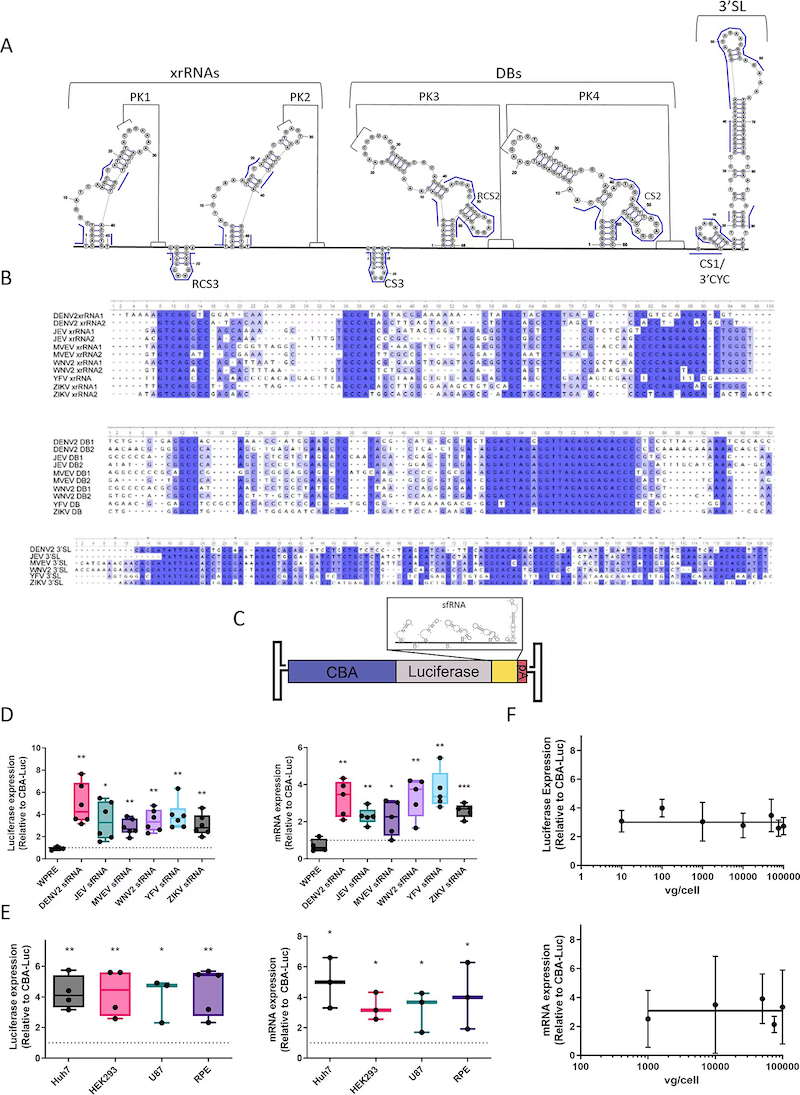
Abstract
Many viruses have evolved structured RNA elements that can influence transcript abundance and translational efficiency, and help evade host immune factors by hijacking cellular machinery during replication. Here, we evaluated the functional impact of sub-genomic flaviviral RNAs (sfRNAs) known to stall exoribonuclease activity, by incorporating these elements into recombinant adeno-associated viral (AAV) genome cassettes. Specifically, sfRNAs from Dengue, Zika, Japanese Encephalitis, Yellow Fever, Murray Valley Encephalitis, and West Nile viruses increased transcript stability and transgene expression compared to a conventional woodchuck hepatitis virus element (WPRE). Further dissection of engineered transcripts revealed that sfRNA elements (i) require incorporation in cis within the 3' untranslated region (UTR) of AAV genomes, (ii) require minimal dumbbell structures to exert the observed effects, and (iii) can stabilize AAV transcripts independent of 5'-3' exoribonuclease 1 (XRN1)-mediated decay. Additionally, preliminary in vivo assessment of AAV vectors bearing sfRNA elements in mice achieved increased transcript abundance and expression in cardiac tissue. Leveraging the functional versatility of engineered viral RNA elements may help improve the potency of AAV vector-based gene therapies.
Importance: Viral RNA elements can hijack host cell machinery to control stability of transcripts and consequently, infection. Studies that help better understand such viral elements can provide insights into antiviral strategies and also potentially leverage these features for therapeutic applications. In this study, by incorporating structured flaviviral RNA elements into recombinant adeno-associated viral (AAV) vector genomes, we show improved AAV transcript stability and transgene expression can be achieved, with implications for gene transfer.
Full Text: https://pmc.ncbi.nlm.nih.gov/articles/PMC11334430/
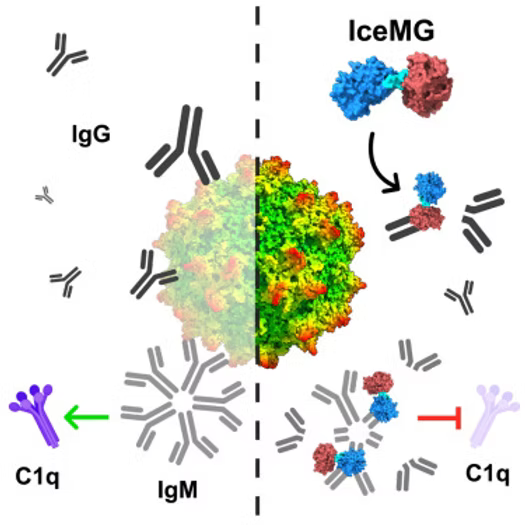
Abstract
Systemic dosing of adeno-associated viral (AAV) vectors poses potential risk of adverse side effects including complement activation triggered by anti-capsid immunity. Due to the multifactorial nature of toxicities observed in this setting, a wide spectrum of immune modulatory regimens are being investigated in the clinic. Here, we discover an IgM cleaving enzyme (IceM) that degrades human IgM, a key trigger in the anti-AAV immune cascade. We then engineer a fusion enzyme (IceMG) with dual proteolytic activity against human IgM and IgG. IceMG cleaves B cell surface antigen receptors and inactivates phospholipase gamma signaling in vitro. Importantly, IceMG is more effective at inhibiting complement activation compared with an IgG cleaving enzyme alone. Upon IV dosing, IceMG rapidly and reversibly clears circulating IgM and IgG in macaques. Antisera from these animals treated with IceMG shows decreased ability to neutralize AAV and activate complement. Consistently, pre-conditioning with IceMG restores AAV transduction in mice passively immunized with human antisera. Thus, IgM cleaving enzymes show promise in simultaneously addressing multiple aspects of anti-AAV immunity mediated by B cells, circulating antibodies and complement. These studies have implications for improving safety of AAV gene therapies and possibly broader applications including organ transplantation and autoimmune diseases.
Full Text: https://www.sciencedirect.com/science/article/pii/S1525001624003058?via%3Dihub
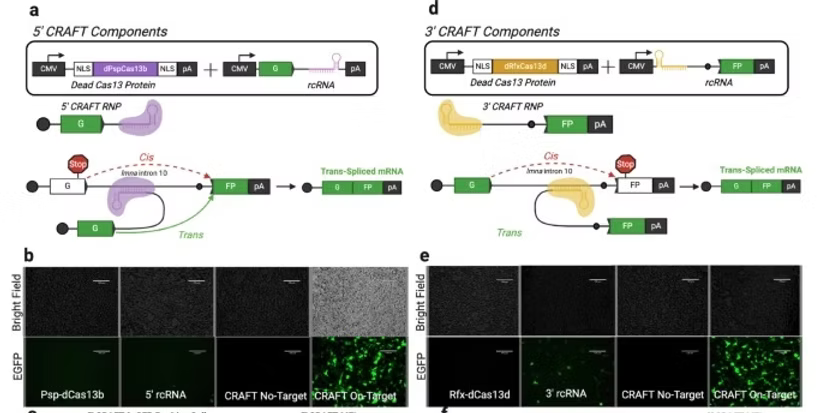
Type VI CRISPR enzymes have been developed as programmable RNA-guided Cas proteins for eukaryotic RNA editing. Notably, Cas13 has been utilized for site-targeted single base edits, demethylation, RNA cleavage or knockdown and alternative splicing. However, the ability to edit large stretches of mRNA transcripts remains a significant challenge. Here, we demonstrate that CRISPR-Cas13 systems can be repurposed to assist trans-splicing of exogenous RNA fragments into an endogenous pre-mRNA transcript, a method termed CRISPR Assisted mRNA Fragment Trans-splicing (CRAFT). Using split reporter-based assays, we evaluate orthogonal Cas13 systems, optimize guide RNA length and screen for optimal trans-splicing site(s) across a range of intronic targets. We achieve markedly improved editing of large 5’ and 3’ segments in different endogenous mRNAs across various mammalian cell types compared to other spliceosome-mediated trans-splicing methods. CRAFT can serve as a versatile platform for attachment of protein tags, studying the impact of multiple mutations/single nucleotide polymorphisms, modification of untranslated regions (UTRs) or replacing large segments of mRNA transcripts.
Full Article: https://www.nature.com/articles/s41467-024-46172-4
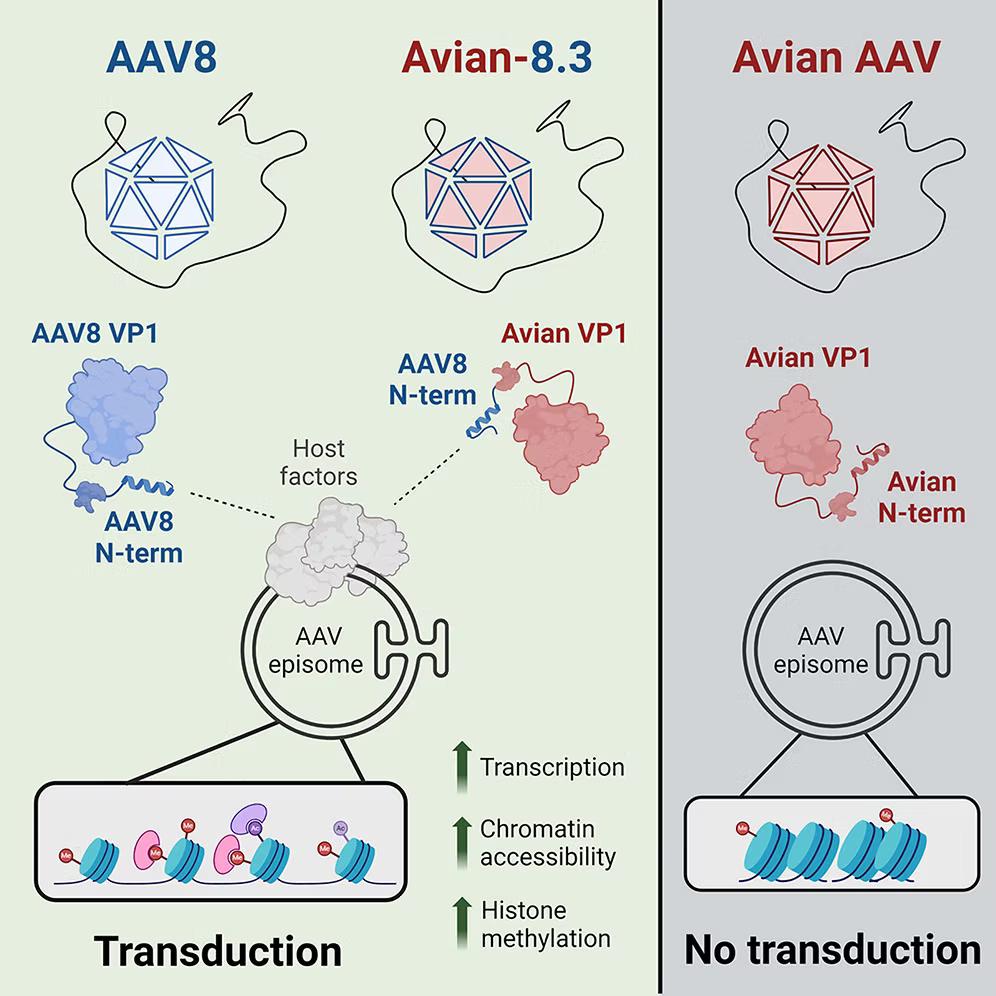
Adeno-associated virus (AAV) is a member of the genus Dependoparvovirus, which infects a wide range of vertebrate species. Here, we observe that, unlike most primate AAV isolates, avian AAV is transcriptionally silenced in human cells. By swapping the VP1 N terminus from primate AAVs (e.g., AAV8) onto non-mammalian isolates (e.g., avian AAV), we identify a minimal component of the AAV capsid that controls viral transcription and unlocks robust transduction in both human cells and mouse tissue. This effect is accompanied by increased AAV genome chromatin accessibility and altered histone methylation. Proximity ligation analysis reveals that host factors are selectively recruited by the VP1 N terminus of AAV8 but not avian AAV. Notably, these include AAV essential factors implicated in the nuclear factor κB pathway, chromatin condensation, and histone methylation. We postulate that the AAV capsid has evolved mechanisms to recruit host factors to its genome, allowing transcriptional activation in a species-specific manner.
Full Article: https://doi.org/10.1016/j.celrep.2024.113902
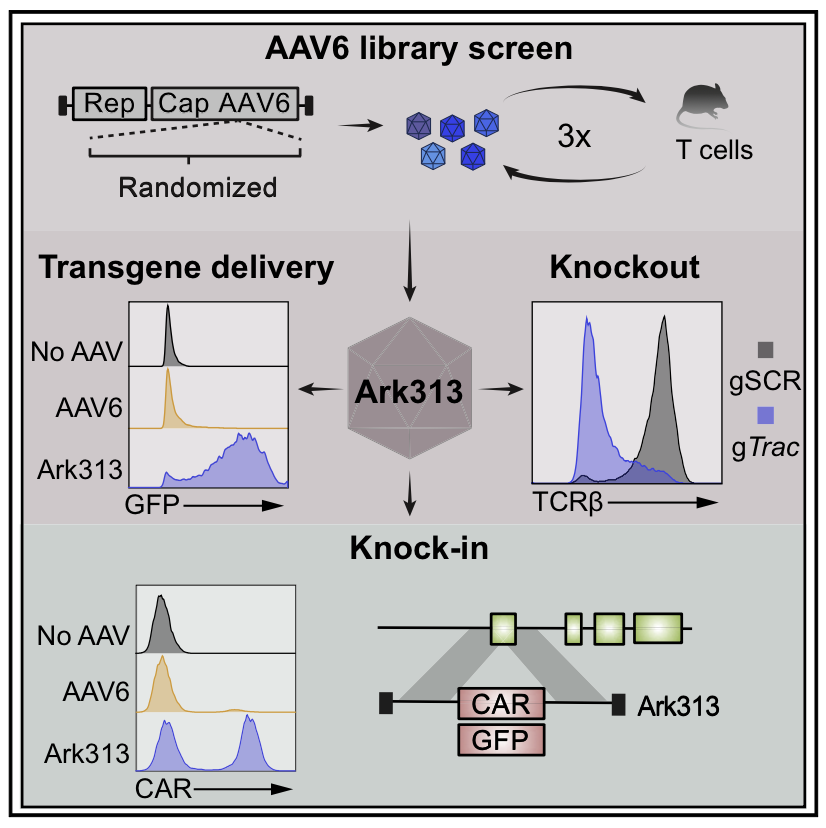
Precise targeting of large transgenes to T cells using homology-directed repair has been transformative for adoptive cell therapies and T cell biology. Delivery of DNA templates via adeno-associated virus (AAV) has greatly improved knockin efficiencies, but the tropism of current AAV serotypes restricts their use to human T cells employed in immunodeficient mouse models. To enable targeted knockins in murine T cells, we evolved Ark313, a synthetic AAV that exhibits high transduction efficiency in murine T cells. We performed a genome-wide knockout screen and identified QA2 as an essential factor for Ark313 infection. We demonstrate that Ark313 can be used for nucleofection-free DNA delivery, CRISPR-Cas9-mediated knockouts, and targeted integration of large transgenes. Ark313 enables preclinical modeling of Trac-targeted CAR-T and transgenic TCR-T cells in immunocompetent models. Efficient gene targeting in murine T cells holds great potential for improved cell therapies and opens avenues in experimental T cell immunology.
Full Article: https://www.sciencedirect.com/science/article/pii/S0092867422015355
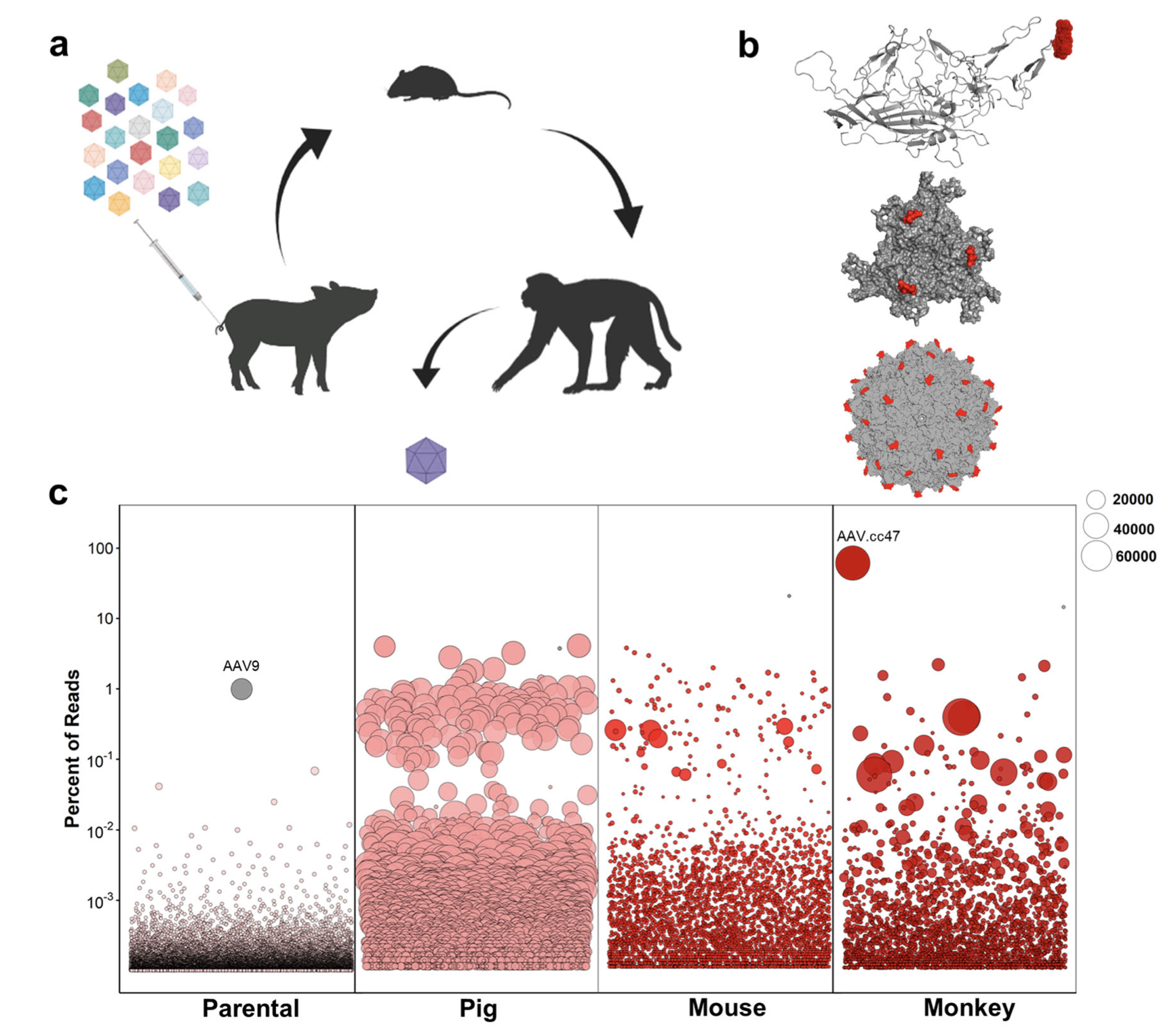
Recombinant adeno-associated viral (AAV) vectors are a promising gene delivery platform, but ongoing clinical trials continue to highlight a relatively narrow therapeutic window. Effective clinical translation is confounded, at least in part, by differences in AAV biology across animal species. Here, we tackle this challenge by sequentially evolving AAV capsid libraries in mice, pigs and macaques. We discover a highly potent, cross-species compatible variant (AAV.cc47) that shows improved attributes benchmarked against AAV ser- otype 9 as evidenced by robust reporter and therapeutic gene expression, Cre recombination and CRISPR genome editing in normal and diseased mouse models. Enhanced transduction efficiency of AAV.cc47 vectors is further cor- roborated in macaques and pigs, providing a strong rationale for potential clinical translation into human gene therapies. We envision that ccAAV vectors may not only improve predictive modeling in preclinical studies, but also clinical translatability by broadening the therapeutic window of AAV based gene therapies.
Full Article: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9548504/pdf/41467_2022_Art…;
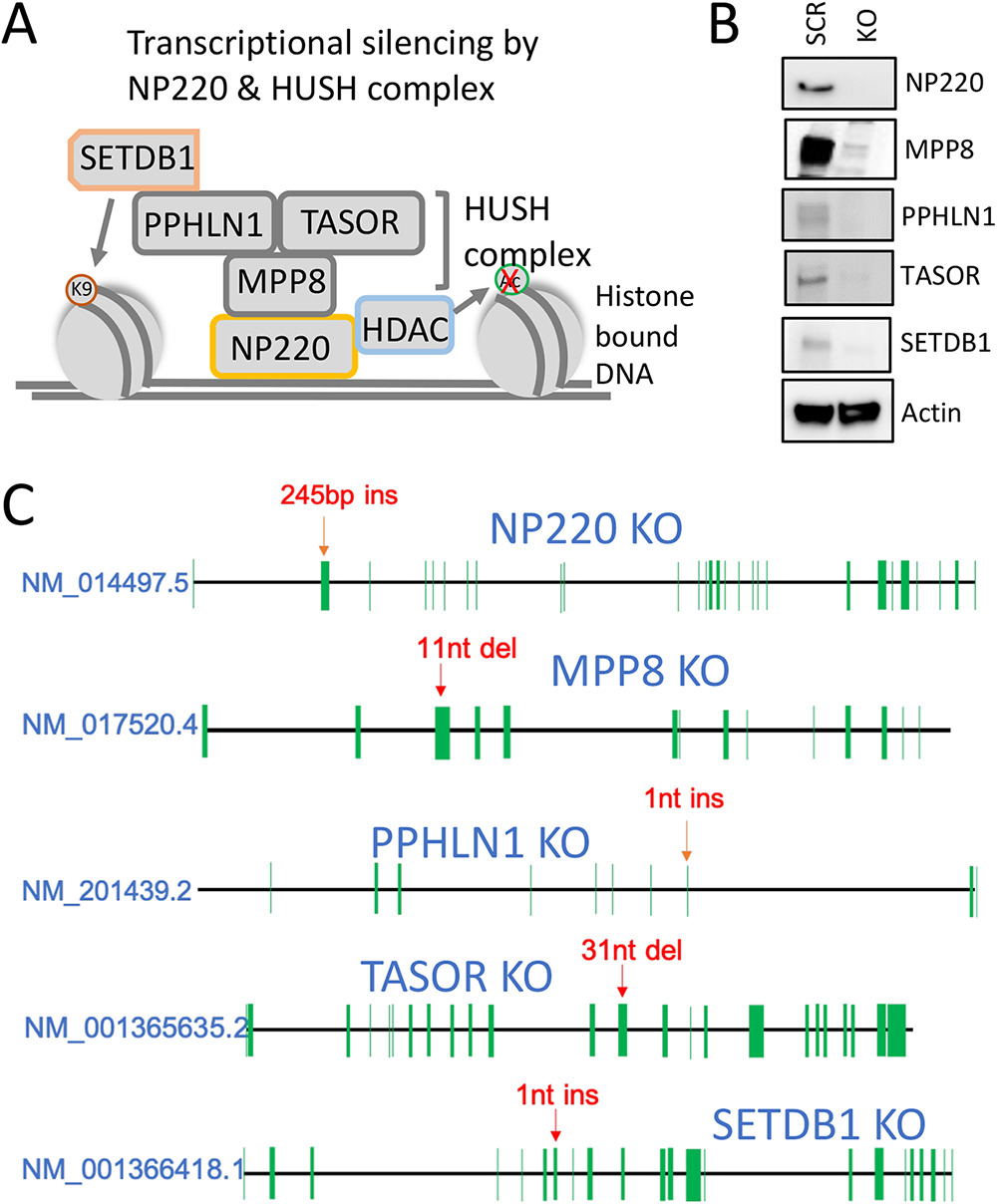
The single-stranded DNA genome of adeno-associated viruses (AAV) undergoes second-strand synthesis and transcription in the host cell nucleus. While wild-type AAV genomes are naturally silenced upon integration into the host genome, recombinant AAV (rAAV) genomes typically provide robust expression of transgenes persisting as extrachromosomal DNA or episomes. Episomal DNA associating with host histones is subject to epigenetic modifications, although the mechanisms underlying such are not well understood. Here, we provide evidence that the double-stranded DNA binding protein NP220, in association with the human silencing hub (HUSH) complex, mediates transcriptional silencing of single-stranded as well as self-complementary rAAV genomes. In cells lacking NP220 or other components of the HUSH complex, AAV genome transcript levels are increased and correlate with a marked reduction in repressive H3K9 histone methylation marks. We also provide evidence that the AAV capsid (serotype) can profoundly influence NP220-mediated silencing of packaged genomes, indicating potential role(s) for capsid-genome or capsid-host factor interactions in regulating epigenetic silencing of rAAV genomes.
Full Article: https://journals.asm.org/doi/full/10.1128/jvi.02039-21?rfr_dat=cr_pub++…
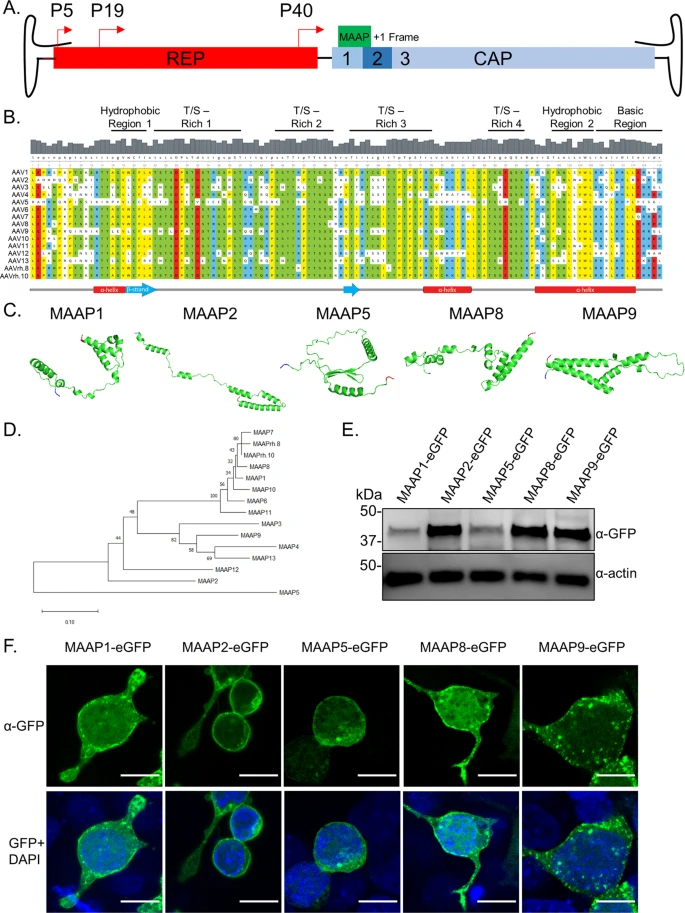
Adeno-associated viruses (AAV) rely on helper viruses to transition from latency to lytic infection. Some AAV serotypes are secreted in a pre-lytic manner as free or extracellular vesicle (EV)-associated particles, although mechanisms underlying such are unknown. Here, we discover that the membrane-associated accessory protein (MAAP), expressed from a frameshifted open reading frame in the AAV cap gene, is a novel viral egress factor. MAAP contains a highly conserved, cationic amphipathic domain critical for AAV secretion. Wild type or recombinant AAV with a mutated MAAP start site (MAAPΔ) show markedly attenuated secretion and correspondingly, increased intracellular retention. Trans-complementation with MAAP restored secretion of multiple AAV/MAAPΔ serotypes. Further, multiple processing and analytical methods corroborate that one plausible mechanism by which MAAP promotes viral egress is through AAV/EV association. In addition to characterizing a novel viral egress factor, we highlight a prospective engineering platform to modulate secretion of AAV vectors or other EV-associated cargo.
Full Article: https://www.nature.com/articles/s41467-021-26485-4
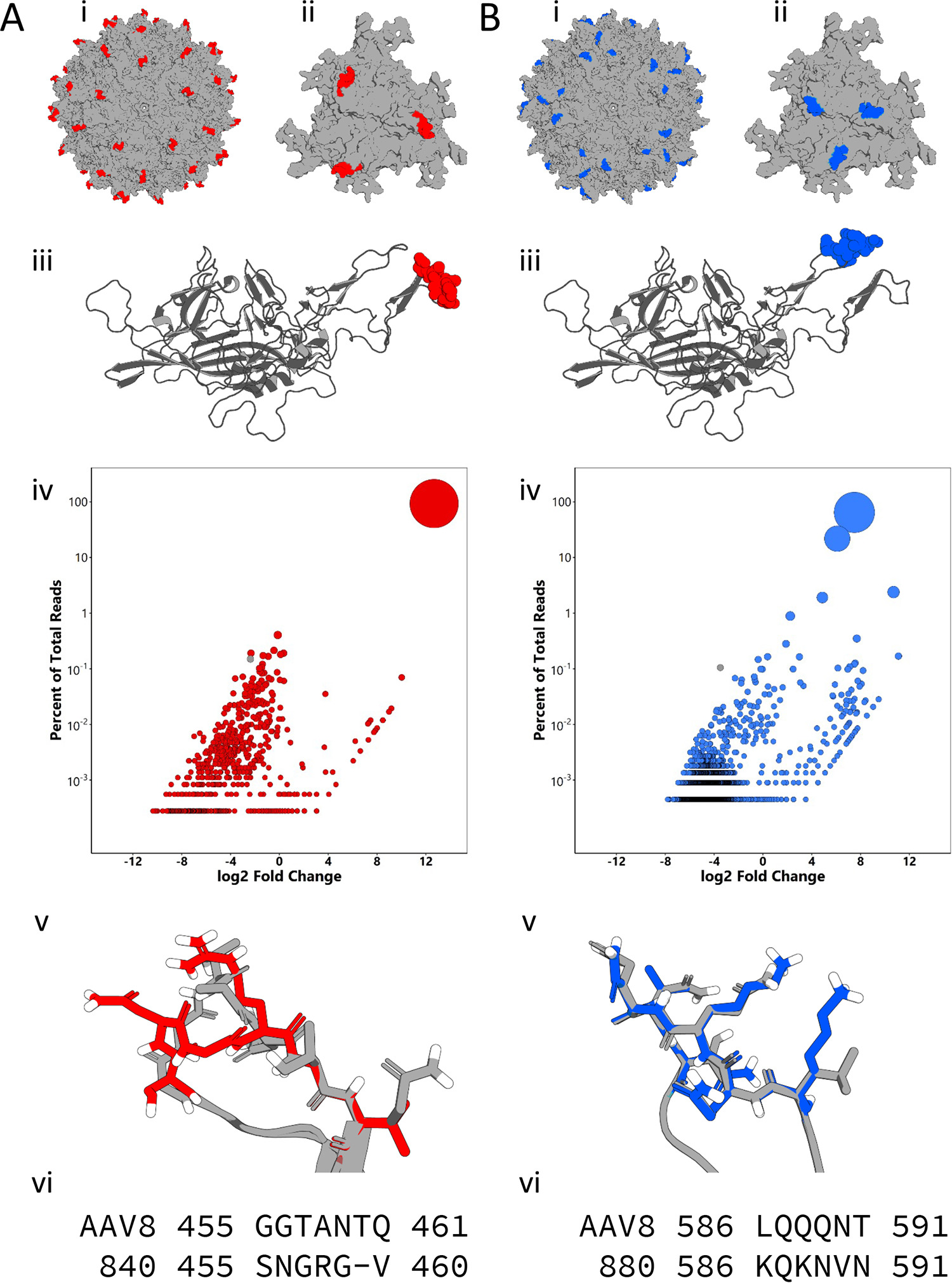
Adeno-associated viruses utilize different glycans and the AAV receptor (AAVR) for cellular attachment and entry. Directed evolution has yielded new AAV variants; however, structure-function correlates underlying their improved transduction are generally overlooked. Here, we report that infectious cycling of structurally diverse AAV surface loop libraries yields functionally distinct variants. Newly evolved variants show enhanced cellular binding, uptake, and transduction, but through distinct mechanisms. Using glycan-based and genome-wide CRISPR knockout screens, we discover that one AAV variant acquires the ability to recognize sulfated glycosaminoglycans, while another displays receptor switching from AAVR to integrin β1 (ITGB1). A previously evolved variant, AAVhum.8, preferentially utilizes the ITGB1 receptor over AAVR. Visualization of the AAVhum.8 capsid by cryoelectron microscopy at 2.49-Å resolution localizes the newly acquired integrin recognition motif adjacent to the AAVR footprint. These observations underscore the new finding that distinct AAV surface epitopes can be evolved to exploit different cellular receptors for enhanced transduction.
Full Article: https://journals.asm.org/doi/full/10.1128/JVI.00587-21?rfr_dat=cr_pub++…
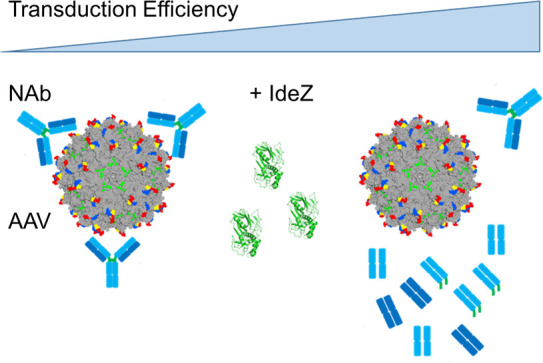
Preexisting humoral immunity to recombinant adeno-associated virus (AAV) vectors restricts the treatable patient population and efficacy of human gene therapies. Approaches to clear neutralizing antibodies (NAbs), such as plasmapheresis and immunosuppression, are either ineffective or cause undesirable side effects. Here, we describe a clinically relevant strategy to rapidly and transiently degrade NAbs before AAV administration using an IgG-degrading enzyme (IdeZ). We demonstrate that recombinant IdeZ efficiently cleaved IgG in dog, monkey, and human antisera. Prophylactically administered IdeZ cleaved circulating human IgG in mice and prevented AAV neutralization in vivo. In macaques, a single intravenous dose of IdeZ rescued AAV transduction by transiently reversing seropositivity. Importantly, IdeZ efficiently cleaved NAbs and rescued AAV transduction in mice passively immunized with individual human donor sera representing a diverse population. Our antibody clearance approach presents a potentially new paradigm for expanding the prospective patient cohort and improving efficacy of AAV gene therapy.
Full Article: https://pubmed.ncbi.nlm.nih.gov/32941184/
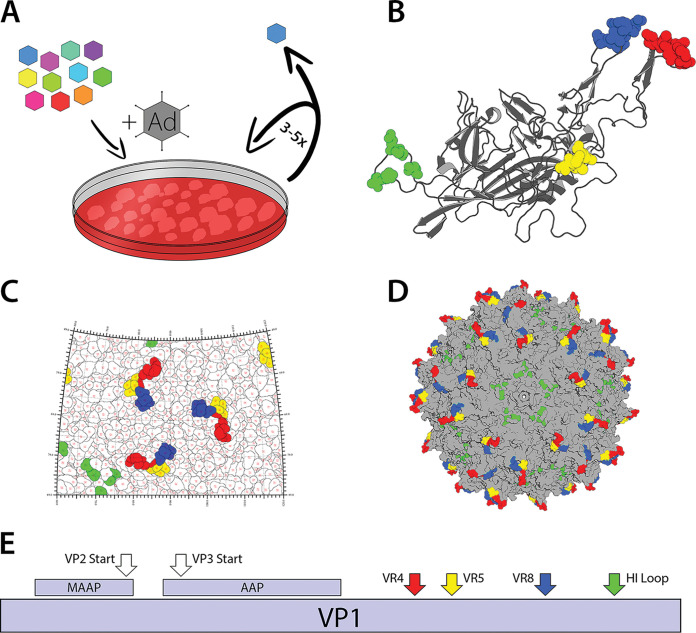
Approaches to engineer recombinant capsids for gene therapy applications have focused on rational design or library-based approaches that can address one or two desirable attributes; however, there is an unmet need to comprehensively improve AAV vector properties. Here, we solve the structures of a natural AAV isolate complexed with antibodies using cryo-electron microscopy and harness this structural information to engineer AAV capsid libraries through saturation mutagenesis of different antigenic footprints. Each surface loop was evolved by infectious cycling in the presence of a helper adenovirus to yield a new AAV variant that then serves as a template for evolving the next surface loop. This stepwise process yielded a humanized AAV8 capsid (AAVhum.8) displaying nonnatural surface loops that simultaneously display tropism for human hepatocytes, increased gene transfer efficiency, and neutralizing antibody evasion. Specifically, AAVhum.8 can better evade neutralizing antisera from multiple species than AAV8. Further, AAVhum.8 displays robust transduction in a human liver xenograft mouse model with expanded tropism for both murine and human hepatocytes. This work supports the hypothesis that critical properties, such as AAV capsid antibody evasion and tropism, can be coevolved by combining rational design and library-based evolution for clinical gene therapy.
Full Article: https://pubmed.ncbi.nlm.nih.gov/32669336/
Mapping structural features of certain AAVs that enable transport across the brain vasculature
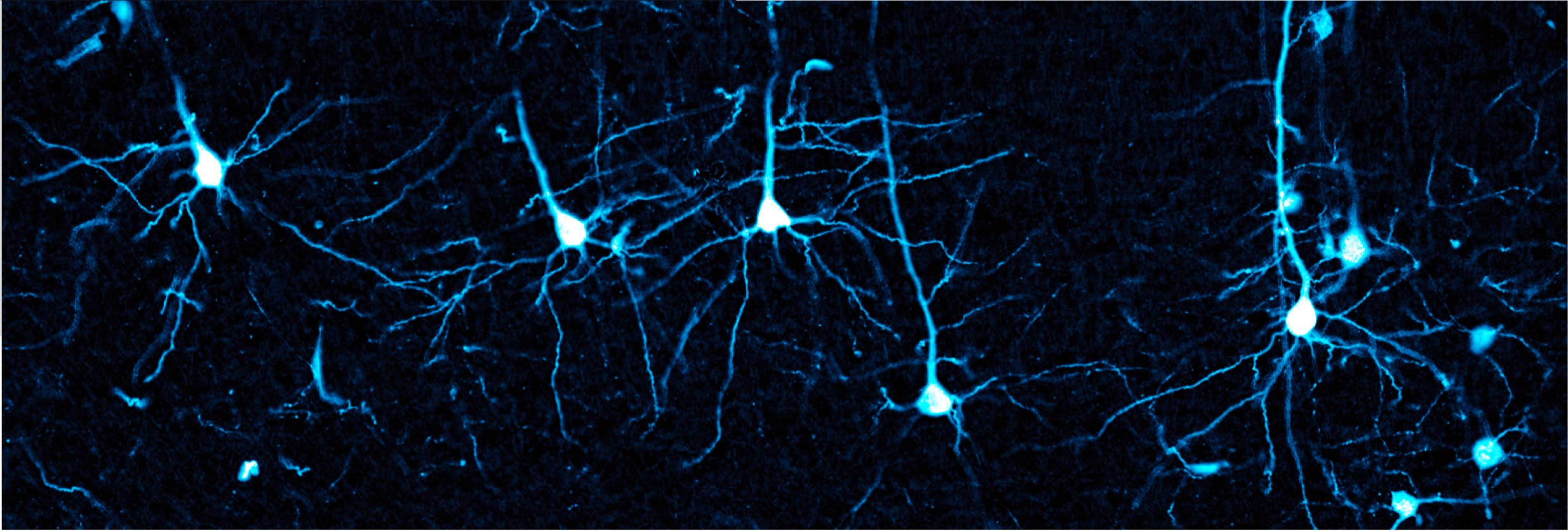
Effective gene delivery to the CNS by intravenously administered adeno-associated virus (AAV) vectors requires crossing the blood-brain barrier (BBB). To achieve therapeutic CNS transgene expression, high systemic vector doses are often required, which poses challenges such as scale-up costs and dose-dependent hepatotoxicity. To improve the specificity and efficiency of CNS gene transfer, a better understanding of the structural features that enable AAV transit across the BBB is needed. We generated a combinatorial domain swap library using AAV1, a serotype that does not traverse the vasculature, and AAVrh.10, which crosses the BBB in mice. We then screened individual variants by phylogenetic and structural analyses and subsequently conducted systemic characterization in mice. Using this approach, we identified key clusters of residues on the AAVrh.10 capsid that enabled transport across the brain vasculature and widespread neuronal transduction in mice. Through rational design, we mapped a minimal footprint from AAVrh.10, which, when grafted onto AAV1, confers the aforementioned CNS phenotype while diminishing vascular and hepatic transduction through an unknown mechanism. Functional mapping of this capsid surface footprint provides a roadmap for engineering synthetic AAV capsids for efficient CNS gene transfer with an improved safety profile.
Full Article: https://pubmed.ncbi.nlm.nih.gov/29175157/
A CRISPR Screen Identifies the Cell Polarity Determinant Crumbs 3 as an Adeno-associated Virus Restriction Factor in Hepatocytes
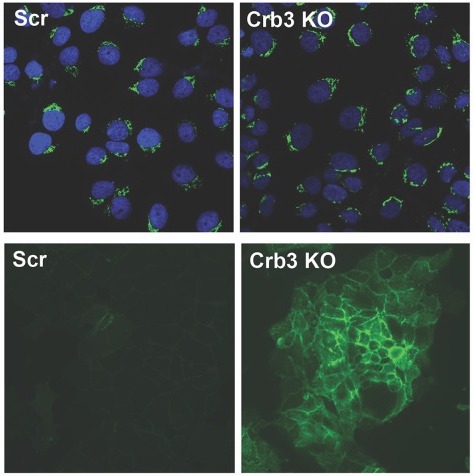
Adeno-associated viruses (AAV) are helper-dependent parvoviruses that have been developed into promising gene therapy vectors. Many studies, including a recent unbiased genomic screen, have identified host factors essential for AAV cell entry, but no genome-wide screens that address inhibitory host factors have been reported. Here, we utilized a novel CRISPR screen to identify AAV restriction factors in a human hepatocyte cell line. The major hit from our gain-of-function screen is the apical polarity determinant Crumbs 3 (Crb3). Knockout (KO) of Crb3 enhances AAV transduction, while overexpression exerts the opposite effect. Further, Crb3 appears to restrict AAV transduction in a serotype- and cell type-specific manner. Particularly, for AAV serotype 9 and a rationally engineered AAV variant, we demonstrate that increased availability of galactosylated glycans on the surfaces of Crb3 KO cells, but not the universal AAV receptor, leads to increased capsid attachment and enhanced transduction. We postulate that Crb3 could serve as a key molecular determinant that restricts the availability of AAV glycan attachment factors on the cell surface by maintaining apical-basal polarity and tight junction integrity
Full article: https://pubmed.ncbi.nlm.nih.gov/31391273/
Modulation of Sialic Acid Dependence Influences the Central Nervous System Transduction Profile of Adeno-associated Viruses
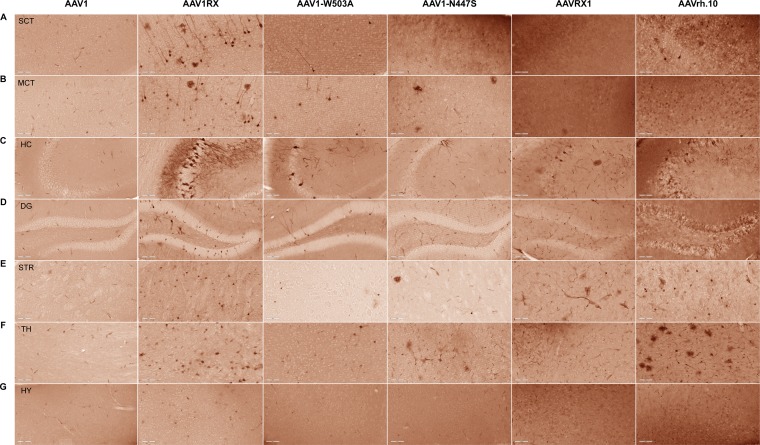
Central nervous system (CNS) transduction by systemically administered recombinant adeno-associated viral (AAV) vectors requires crossing the blood-brain barrier (BBB). We recently mapped a structural footprint on the AAVrh.10 capsid, which, when grafted onto the AAV1 capsid (AAV1RX), enables viral transport across the BBB; however, the underlying mechanisms remain unknown. Here, we establish through structural modeling that this footprint overlaps in part the sialic acid (SIA) footprint on AAV1. We hypothesized that altered SIA-capsid interactions may influence the ability of AAV1RX to transduce the CNS. Using AAV1 variants with altered SIA footprints, we map functional attributes of these capsids to their relative SIA dependence. Specifically, capsids with ablated SIA binding can penetrate and transduce the CNS with low to moderate efficiency. In contrast, AAV1 shows strong SIA dependency and does not transduce the CNS after systemic administration and, instead, transduces the vasculature and the liver. The AAV1RX variant, which shows an intermediate SIA binding phenotype, effectively enters the brain parenchyma and transduces neurons at levels comparable to the level of AAVrh.10. In corollary, the reciprocal swap of the AAV1RX footprint onto AAVrh.10 (AAVRX1) attenuated CNS transduction relative to that of AAVrh.10. We conclude that the composition of residues within the capsid variable region 1 (VR1) of AAV1 and AAVrh.10 profoundly influences tropism, with altered SIA interactions playing a partial role in this phenotype. Further, we postulate a Goldilocks model, wherein optimal glycan interactions can influence the CNS transduction profile of AAV capsids.
Ring finger protein 121 is a potent regulator of adeno-associated viral genome transcription
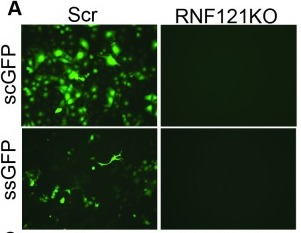
Adeno-associated viruses (AAV) are Dependoparvoviruses that have shown promise as recombinant vectors for gene therapy. While infectious pathways of AAV are well studied, gaps remain in our understanding of host factors affecting vector genome expression. Here, we map the role of ring finger protein 121 (RNF121), an E3 ubiquitin ligase, as a key regulator of AAV genome transcription. CRISPR-mediated knockout of RNF121 (RNF121 KO) in different cells markedly decreased AAV transduction regardless of capsid serotype or vector dose. Recombinant AAV transduction is partially rescued by overexpressing RNF121, but not by co-infection with helper Adenovirus. Major steps in the AAV infectious pathway including cell surface binding, cellular uptake, nuclear entry, capsid uncoating and second strand synthesis are unaffected. While gene expression from transfected plasmids or AAV genomes is unaffected, mRNA synthesis from AAV capsid-associated genomes is markedly decreased in RNF121 KO cells. These observations were attributed to transcriptional arrest as corroborated by RNAPol-ChIP and mRNA half-life measurements. Although AAV capsid proteins do not appear to be direct substrates of RNF121, the catalytic domain of the E3 ligase appears essential. Inhibition of ubiquitin-proteasome pathways revealed that blocking Valosin Containing Protein (VCP/p97), which targets substrates to the proteasome, can selectively and completely restore AAV-mediated transgene expression in RNF121 KO cells. Expanding on this finding, transcriptomic and proteomic analysis revealed that the catalytic subunit of DNA PK (DNAPK-Cs), a known activator of VCP, is upregulated in RNF121 KO cells and that the DNA damage machinery is enriched at sites of stalled AAV genome transcription. We postulate that a network of RNF121, VCP and DNA damage response elements function together to regulate transcriptional silencing and/or activation of AAV vector genomes.
Tissue-Dependent Expression and Translation of Circular RNAs with Recombinant AAV Vectors In Vivo
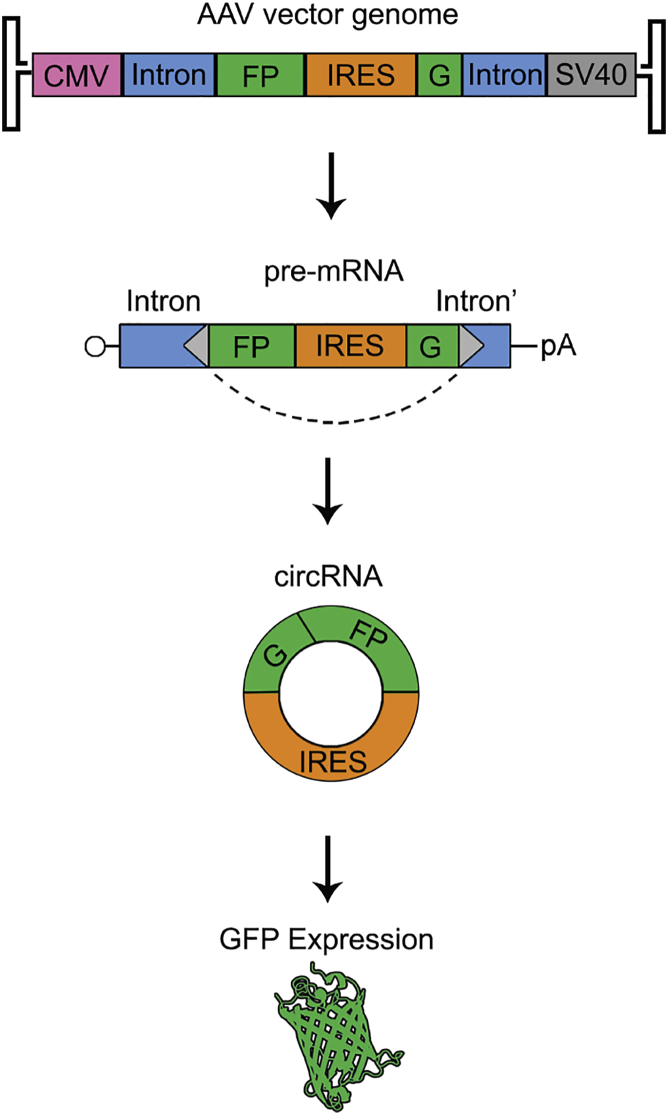
Circular RNAs (circRNAs) are long-lived, covalently closed RNAs that are abundantly expressed and evolutionarily conserved across eukaryotes. Possible functions ranging from microRNA (miRNA) and RNA binding protein sponges to regulators of transcription and translation have been proposed. Here we describe the design and characterization of recombinant adeno-associated viral (AAV) vectors packaging transgene cassettes containing intronic sequences that promote backsplicing to generate circularized RNA transcripts. Using a split GFP transgene, we demonstrate the capacity of vectors containing different flanking intronic sequences to efficiently drive persistent circRNA formation in vitro. Further, translation from circRNA is efficiently driven by an internal ribosomal entry site (IRES). Upon injecting AAV vectors encoding circRNA in mice, we observed robust transgene expression in the heart, but low transduction in the liver for the intronic elements tested. These results highlight the potential for exploiting AAV-based circRNA expression to study circRNA function and tissue-specific regulation in animal models, as well as development of therapeutic platforms using this approach.
Full Article: https://pubmed.ncbi.nlm.nih.gov/30245471/
Lab Members
Current Members
- Abigail Benkert, Cardiothoracic Surgery Resident
- Mourya Durgam Jayaram, Sr. Research Technician
- Jacob Schaff, Research Technician III
- Jie Vanderley, Research Technician III
- Roopali Shrivastava, Scientific/Research Lab Manager
- Garth Devlin, Scientific Program Leader I
- David Fiflis, Postdoc
- Delaney Fisher, Postdoc
- Ezra Loeb, Postdoc
- Harshitha Venugopal Lavanya, Grad Student
- Mark Ochoa, Grad Student
- Xinlong Wan, Grad Student
- Nancy Wang, Grad Student
- Mitch Yip, Grad Student
- Vivian Yudistyra, Grad Student
Gene Therapy Resource Core
The Duke Gene Therapy Resource Core (GTRC) is committed to providing scalable production and analytics for platform AAV & RNA technologies to enable pre-clinical gene therapy development.